Syed Muhammad Ashhad Halimi1 ![]() ,
Muhammad Saeed1,
Safiullah .1,
Khalid Muhammed Khan2
,
Muhammad Saeed1,
Safiullah .1,
Khalid Muhammed Khan2
For correspondence:- Syed Halimi Email: ahhadhalimi@upesh.edu.pk Tel:+92919216750
Received: 4 August 2016 Accepted: 5 January 2017 Published: 26 February 2017
Citation: Halimi SM, Saeed M, . S, Khan KM. Pharmacological evaluation of novel dimers of an arylpropionic acid class of non-selective cyclooxygenase inhibitors. Trop J Pharm Res 2017; 16(2):327-336 doi: 10.4314/tjpr.v16i2.10
© 2017 The authors.
This is an Open Access article that uses a funding model which does not charge readers or their institutions for access and distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0) and the Budapest Open Access Initiative (http://www.budapestopenaccessinitiative.org/read), which permit unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited..
Purpose: To explore and identify cyclooxygenase (COX) inhibitors with optimal potency and efficacy using an arylpropionic acid class of drugs as lead molecules.
Methods: The selected lead molecules were dimerised through chemical processes (reflux condensation) and characterised in terms of structural properties using infrared, proton nuclear magnetic resonance, electron impact mass spectrometry, and elemental analysis techniques. The molecules were evaluated pharmacologically for acute toxicity and anti-inflammatory (carrageenan-induced paw oedema test), analgesic (acetic acid-induced writhing test in mice), and antipyretic (Brewer’s yeast-induced pyrexia test in mice) activities against control (normal saline) and relevant reference standard drugs. Docking analyses were also performed to assess possible protein–ligand interactions.
Results: The test compounds were non-toxic at doses of 50, 100 and 150 mg/kg body weight, ip. Pharmacological evaluation revealed that the test compounds, TC-I through TC-IV, had significant anti-inflammatory and peripheral analgesic activities (p < 0.001). An antipyretic test showed that TC-I, -II, and -III showed highly significant antipyretic activities at all doses tested. TC-IV at 20 and 30 mg/kg body weight exhibited significant antipyretic activities (p < 0.05), while at 50 mg/kg body weight, the activity was highly significant (p < 0.001). Molecular modelling revealed strong inhibitory interactions with docking scores of 116.2, 128.8, 144.2, and 136.0 kcal/mol, respectively, in comparison with the reference ligand, flurbiprofen (94.9 kcal/mol).
Conclusion: The dimerised lead drug molecules showed significant anti-inflammatory, analgesic, and antipyretic activities in animals and may further be explored as potential new drug candidates for inflammatory conditions.
Introduction
Cyclooxygenase (COX) inhibitors, non-steroidal anti-inflammatory drugs (NSAIDs), are commonly prescribed and used for their anti-inflammatory, analgesic, and antipyretic activities. A diverse collection of NSAIDs, such as arylpropionic acids, arylacetic acids, βketoenols, and diary heterocycles, acts by inhibiting the activity of cyclooxygenase (COX) enzymes [1]. NSAID selection depends on the clinical condition of the patient under treatment, COX1 and COX2 selectivity, duration of use, and gastric ulcerative and cardiovascular risks [2].
A prolific area of research looks at existing drug molecules that may be further amplified in terms of potency, efficacy, selectivity, or safety. The development and successful testing of molecular computations for both the receptors (proteins) and ligands (drugs) has further aided the efforts of researchers, to the point of designing appropriate drug structures for both known and unknown proteins, receptors, or enzymes [3,4].
Well-established lead NSAID molecules for synthesising derivatives have the advantages of predicable pharmacological activities, known doses, durations of action, and side and toxic effects. The pharmacological evaluation of such synthesised analogues, in vivo and through computer docking, can provide useful data for further augmentation of the existing lead molecule; for more benefits and fewer side effects, the ‘better’ structure could be screened out among the synthesised analogues for further optimisation of its pharmacological activity. Several success stories have been reported to date regarding such computer-aided drug discoveries [5].
Naproxen [6], flurbiprofen [7], and ibuprofen [8], belonging to the arylpropionic acid class of non-selective COX inhibitors, were selected for synthesis of their dimers and further exploration of these dimers for anti-inflammatory, analgesic, and antipyretic activities. Molecular docking analyses were also performed to assess probable drug-receptor interactions with this new COX inhibitor approach.
Methods
Chemicals
Pharmaceutical-grade ibuprofen, flurbiprofen, and naproxen were provided by Polyfine Chemipharma (Pvt.) Ltd, Hayatabad, Peshawar, Pakistan. Ethanol and concentrated sulphuric acid were obtained from Sigma-Aldrich, USA. Lambda carrageenan (Sigma, USA), glacial acetic acid (Panreac, Spain), Brewer’s yeast (Merck, Germany), and sterile normal saline I/V (Otsoka Japan) were used as received.
The purity of the products was checked on thin-layer chromatographic plates of aluminium coated with silica gel (Kieselgel 60 F25, Merck, Germany). Melting points were assessed with a Bicot apparatus (Bibby Scientific Ltd., UK). Proton nuclear magnetic resonance (1H NMR) measurements were performed using a Bruker AV 300 system (400 MHz) with dimethyl sulphoxide (DMSO) as the solvent. Electron impact mass spectroscopy (EI MS) was conducted with a JEOL MS Route spectrometer (JMS 600-H). Infrared (IR) spectra were obtained with a Jasco IR-A-302 spectrometer. Ultraviolet (UV) spectra were generated with a Thermo Electron Vision Pro (ver. 4.10). C:H:N analyses were performed with a Carlo ErbaStrumentazione system (Mod-1106, Italy).
Anti-inflammatory activity tests were conducted using a plethysmometer (Model LE 7500 Plan lab S.L.). Antinociceptive activity was assessed with a hot plate analgesiometer (Harvard Apparatus, USA). Antipyretic activity was measured using a digital thermometer (Model CA92121, ACON Laboratories, USA).
Synthesis
Ethyl ester analogues of naproxen, flurbiprofen, and ibuprofen were synthesised through reflux condensation of the reactants in an equimolar (0.1 mmol:0.1 mmol) ratio in an acidic environment, with the addition of a few drops of concentrated sulphuric acid to the reaction [9]. Completion of the reaction was monitored using thin layer chromatography (TLC), with ethyl acetate and hexane as the mobile phase. The reaction duration ranged from 12–18 h. The resulting solid products were filtered, washed with hot hexane (200–400 mL) and distilled water (400–500 mL), and then dried (Step 1). Esterified compounds thus synthesised were treated with hydrazine hydrate, and again subjected to reflux condensation for 24–48 h, using ethanol as the solvent [10]. Completion of the reactions was ascertained using TLC. The products were then purified with different techniques (Step 2). The esters and hydrazides thus synthesised were refluxed overnight, in the presence of carbonyldiimidazole (CDI) as the catalyst, using acetonitrile as the reaction medium. General Scheme I shows dimer formation in a three-step reaction. The products formed were then purified similarly through the different techniques.
Experimental animals
Mice (BALB-c) from the National Institute of Health (NIH) Islamabad, Pakistan, were used for the experimental studies. Mice were fed with standard food ad libitum with free access to drinking water, and kept at 25 ± 2°C with a 12-h/12-h day/night cycle. The animal studies were approved by the Ethical Committee of the University of Peshawar, Pakistan (ref no. 01/EC- 15/Pharm), according to international guidelines for the handling of animals [11,12].
Acute toxicity test
For the toxicity study, 18 mice were distributed randomly into three groups, n = 6 each. Groups were treated with 50, 100, and 500 mg/kg bodyweight of the test compounds intraperitoneally (ip) through injection. The animals were observed for 6 h for any behavioural changes; mortality rates were determined at 24 h after administration of the test compounds [13].
Anti-inflammatory activity (carrageenan-induced paw oedema) test
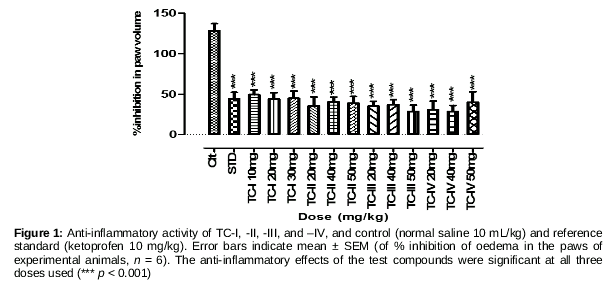
This test was performed on 30 mice distributed randomly into five groups, n = 6 each. Inflammation was induced by injecting 0.1 mL of 1% carrageenan solution into the plantar surface of the mouse hind paw [14]. Test groups I, II, and III were treated with 10, 20, and 30 mg/kg of the test compound, respectively. Group IV (control) was treated with normal saline (1 mL/kg) while group V (reference standard) was treated with ketoprofen (10 mg/kg), 30 min before carrageenan injection. The paw volume was measured at 0, 1, 2, 3, 4, and 5 h using a digital plethysmometer. The difference between the readings at time 0 h and the different time points was taken as the degree of oedema, quantified as
% Inhibition = (α – β)/α × 100……………. (1)
where α and β indicate the increase in paw volume of control and drug-treated animals, respectively.
Evaluation of anti-nociceptive activity
Acetic acid-induced writhing test in mice
Mice were distributed into five groups [groups I, II, III, IV (control), and V (reference standard)], n = 6 each; at levels of 10, 20, and 30 mg/kg body weight ip, doses of the test compounds were administered to the animals of the first three groups I–III, respectively. The animals of group IV received normal saline (10 mL/kg body weight, ip) while those of group V were treated with diclofenac at a dose of 10 mg/kg body weight, ip. After an interval of 30 min after treatment, all groups were treated with 0.6% acetic acid at 10 mL/kg body weight, ip [15]. The mice were then placed in individual cages and the numbers of writhes were counted for each mouse for a period of 30 min, with a 5-min latency period.
Hot plate (thermal) test in mice
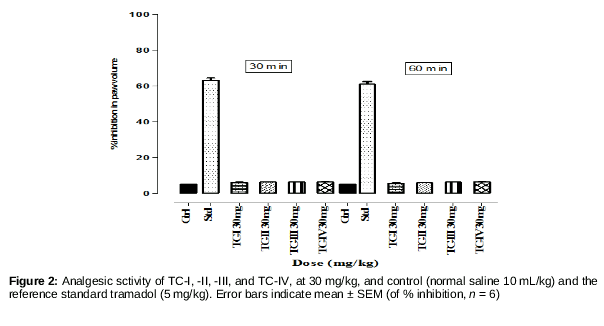
Mice weighing 18–22 g of both genders were used. As described in a reported protocol, animals habituated to the laboratory environment for 2 h were distributed into five groups, n = 6 each. Test groups I, II, and III were treated with 10, 20, and 30 mg/kg/ip, respectively, of the test compound; group IV was treated with normal saline (10 mL/kg, ip); and group V with tramadol at 5 mg/kg, ip. The animals were subjected to the hot plate test after 30- and 60-min intervals; the hot plate was maintained at a temperature of 54.0 ± 0.1°C. The latency time (s), i.e., the reaction to the hot plate, was recorded by noting the reaction of the experimental animal, licking, flicking of the hind limb, or jumping from the cylinder [16]. The cut-off time was fixed at 30 s for the animal’s reaction to avoid tissue damage due to prolonged exposure to the hot plate. Anti-nociception was determined using the formula:
% Anti-nociception = [(t-c)/(s-c)] × 100 …….. (2)
where t is the test latency, c is the control latency, and s is the cut-off time.
Anti-pyretic study (Brewer’s yeast-induced pyrexia test)
Mice of either sex, weighing 25 – 30 g, were used. As described in a reported protocol, before the experimental procedure, the animals fasted overnight but with free access to water. The animals were distributed into five groups, n = 6 each. Doses of 10, 20, and 30 mg/kg of the test compound were administered to the animals of the first three groups (groups I–III), respectively. Animals of group IV were treated with normal saline (10 mL/kg), and those of group V were treated with paracetamol (150 mg/kg). Brewer’s yeast solution (20 %) was administered subcutaneously (sc) below the nape of neck in the back of the mice (10 mL/kg body weight) to induce hyperpyrexia in the experimental animals in all groups [17]. Twenty-four hours after the injection, changes in rectal temperature were recorded by inserting an oil-lubricated digital thermometer into their rectums at 1, 2, 3, 4, and 5 h. Only those mice whose rectal temperatures had increased by at least 0.3–0.5 °C were selected for further test procedures.
Statistical analysis
For statistical analyses, GraphPad Prism (ver. 5.01) software was used. Data pertaining to pharmacological evaluation were analysed by applying one-way analysis of variance (ANOVA) followed by Dunnett’s post hoc analysis. P < 0.001 was considered to be highly significant while p < 0.05 was considered statistically significant in this study.
Molecular docking simulation analysis
AutoDock (ver. 4.0.1.) software was used for docking simulation analyses. A rigid-protein-and-flexible-ligand standard docking procedure was performed with the AutoDock Tools, using a Lamarckian generic logarithm. A three-dimensional 60 × 60 × 60 point grid was built, centred on the centre of the M loop of COX2 (PDB ID: 3PGH). An energy map was calculated using a grid spacing of 0.375 Å and a distance-dependent function of the dielectric constant, while default settings were used for the other parameters. After completion of the docking, the most favourable free binding energies were selected as the resulting ligand–protein interactions [18]. Flurbiprofen was used as a reference and co-crystallised ligand was used to assess binding mode accuracy and test compound affinity [19].
Results
Chemistry of the synthesised dimers
Test compound (TC)-I: mp = semisolid; yield: 0.389 g (82.67%): IR (KBr): nmax 3,527.6, 3,144.7, 2,826.5, 1,964.4, 1,807.2, 1,624.9, 1,483.2, 1,414.7, 1,341.4, 1,133.1, 939.3, 914.2, 769.5, 701.1 cm−1; 1H NMR (400 MHz, chloroform): δ 10.08 (s, 1H), 7.77–7.66 (m, 3H), 7.55–7.38 (m, 7H), 7.36–7.27 (m, J = 12.0 Hz, 2H), 7.23 (t, J = 1.2, 1.6 Hz 1H), 7.06 (dd, J = 7.6, 1.6, 1H), 3.81 (s, 3H), 3.52 (m, 2H), 1.34 (d, 1H), 1.28 (d, 1H). EI MS: m/z (relative abundance %): 472 (M+, 5.7), 360 (7.3), 300 (28.3), 256 (33.2), 220 (100) 156 (60), 136 (30), 80 (10); elemental analysis for C29H27FN2O3: C, 72.91; H, 5.28; F, 5.24; N, 7.73; O, 8.83.
Test compound (TC)-II: mp = liquid; yield: 0.28 g (67.4%): IR (KBr): nmax 3,160, 2,955, 1,781, 1,624, 1,512, 1,459, 1,208, 924, 847, 734 cm−1; 1H NMR (300 MHz, DMSO-d6): δ 12.16 (s, 2H, NH), 7.14 (m, 8H, H-2/3/5/6), 4.07 (q, 2H, CH), 2.39 (d, 4H, CH2), 1.78 (m, 2H, CH), 1.47 (d, 6H, CH3), 0.83 (d, 12H, CH3); EI MS: m/z (relative abundance %): 246 (50.9), 203 (100), 118 (49.7), 91 (26.4), 43 (7.5); elemental analysis for C26H36N2O2: C, 76.43; H, 8.88; N, 6.86; found: C, 75.98; H, 8.62; N, 6.90.
Test compound (TC)-III: MP: 77.35°C; yield: 0.24 g, 53.73%: IR (KBr): nmax 3,143, 1,770, 1,604, 1,504, 1,229, 1,031, 960, 820, 745, 719 cm−1; 1H NMR (300 MHz, DMSO-d6): δ 12.16 (s, 2H, NH), 7.80 (m, 4H, H-4/8), 7.73 (d, j1,3 = 0.5 Hz, 2H, H-1), 7.37 (dd, j3, 4 = 8.5 Hz, j3, 1 = 2.0 Hz, 2H, H-3), 7.30 (d, j5,7 = 2.5 Hz, 2H, H-5), 7.16 (dd, j7, 8 = 9.0 Hz, j7, 5 = 2.5 Hz, 2H, H-7), 4,25 (q, 2H, CH), 3.30 (s, 6H, OCH3), 1.56 (d, 6H, CH3); EI MS: m/z (relative abundance %): 456 (M+, 3.4), 438 (58.1), 270 (33.3), 212 (57), 185 (100); elemental analysis for C28H28N2O4: C, 73.66; H, 6.18; N, 6.14; found: C, 73.68; H, 5.17; N, 6.19.
Test compound (TC)-IV: mp: 185.5°C, Yield: 0.38 g, 79.5%: IR (KBr): nmax 3,145, 2,826, 1,806, 1,762, 1,624, 1,483, 1,414, 1,341, 939, 769 cm−1; 1H NMR (300 MHz, DMSO-d6): δ 11.32 (s, 2H, NH), 7.44 (m, 12H, H-5/6/2´/3´/5´/6´), 7.25 (m, 4H, H-2/4´), 4.23 (q, 2H, CH), 1.53 (d, 6H, CH3); EI MS: m/z (relative abundance %): (M+, absent), 466 (3.9), 284 (100), 269 (63.6), 225 (27.7), 43 (43.4); elemental analysis for C30H26F2N2O2: C, 74.36; H, 5.41; N, 5.78; found: C, 74.28; H, 5.82; N, 5.84.
Acute toxicity
No acute toxicity was observed over the 24 h following dosing of 50, 100, or 150 mg/kg body weight of any test compound.
Anti-inflammatory activity
Anti-inflammatory activity data were analysed using one-way ANOVA followed by Dunnett’s post hoc analysis. Figure 1 shows that all test compounds had statistically significant anti-inflammatory effects (*** p < 0.0001) at the given doses with reference to normal saline (10 mL/kg) as a control and ketoprofen 10 mg/kg as a reference standard. Doses of the four compounds were selected on the basis of average doses of the lead drug molecules used.
Anti-nociceptive activity
The hot plate test was negative for all of the test compounds, indicating that the test compounds had no central analgesic activity. To avoid a congested presentation, data for only the 30 mg doses are reported. The overall result of this test was negative.
Data for the acetic acid-induced writing test indicated that the test compounds (TC-I, -II ‑III, and -IV) were active versus the normal saline 10 mL/kg control and diclofenac 10 mg/kg reference standard (*** p < 0.001), indicating a clear dose–response relationship ().
Anti-pyretic activity
Brewer’s yeast-induced pyrexia test results showed that the antipyretic activity of the four compounds was significant (p < 0.001) at all of the doses tested versus the control (normal saline 10 mL/kg) and paracetamol (150 mg/kg body weight) reference standard (Figure 4).
Molecular docking results
Molecular docking simulates and determines the best-fit orientation of the ligand (drug) on the basis of binding scores (energies), at the specific binding site of the receptor protein, which can authenticate or suggest pharmacological activity and drug design rationales, with reference to the receptor protein studied [20]. COX1, a constitutively expressed enzyme, catalyses primarily the production of prostaglandins that regulate homeostasis, platelet activity, and gastric and renal function. COX-2, is a constitutive and inducible enzyme that catalyses predominantly the production of proinflammatory prostaglandins [21].
Computational modelling tests with the TCs revealed that the ‘best’ scoring conformations (energies) of the TCs (I, II, III, and IV) were much higher than that of flurbiprofen, the reference ligand (, Figures. 5–9). Superimposition of the most favourable conformations of the test compounds and of the reference ligand (Figure 10) showed that they were accommodated well within the COX2 active site pocket.
Discussion
Despite remarkable advances in pharmaceutical chemistry, inflammatory conditions remain major challenges in terms of both curative and symptomatic treatments. More effective and safer COX inhibitors are required. In our present study featuring pharmacological and computational analysis, we discovered an interesting aspect of COX targeting. What we term “dimerised COX inhibitors” are more potent than are lead molecules. All test compounds exhibited highly significant anti-inflammatory and peripheral analgesic activities, and such activities were dose-dependent. The effectiveness of an NSIAD is related to the rapidity and efficacy of access through the narrow COX channel created by Arg-120, Tyr-355, and Glu-524 [22]. Our in vivo tests clearly showed that COX2 was effectively inhibited by dimerised analogues of selective lead molecules that were larger in size than unitary lead molecules. The active site of COX2, which encompasses the Val-523 amino acid residue, permits secondary, internal hydrophobic extension of the binding site to accommodate larger molecules [23]. We suggest that this explains the responses to our test compounds; this suggestion is supported by our molecular docking analysis. The docking scores of the test compounds (), and superimposition of the most favourable confirmations [24] (0), both support this hypothesis. Arylpropionic acid COX inhibitors bind in one of two reported poses within the COX binding pocket: (i) in a way that the carboxylate moiety is bound to the edifice site residues Arg-120 and Tyr-355, such as flurbiprofen [25, 26], or (ii) whereby the carboxylate moiety binds to the side chains of Tyr-385 and/or Ser-530 residues in an upturned fashion, such as diclofenac [20]. The ‘best’ conformations of the test compounds showed that TC-I was bound via a hydrogen bond with Ser-530, TC-II with Try-355, TC-III had only hydrophobic interactions (Val-116, Leu-359, Leu‑351, Leu-354, Val-344, Leu-352, Phe-518, Leu-384, Val-523, and Met-522), also reported as a ‘second anchoring site’ by Jelena [23]. In contrast, TC-IV featured a hydrogen bond between Try-385 and Ser‑530. Computational studies firmly supported the data of the in vivo experiments. It appears that the dimerisation concept may be used to identify useful lead compounds, opening a novel window into the management of overwhelming inflammatory disorders. These fascinating detailed findings warrant further mechanistic studies, with a focus on sequential virtual design, chemical modification, and 3DQSAR evaluation. Marketable therapies may result.
Conclusion
The in vivo results demonstrate that dimerisation, homo- or hetero-, within the selected lead drug molecules increased pharmacological activities (anti-inflammatory, analgesic and antipyretic). Thus, all four dimers should be assessed further as new candidate molecules for the treatment of inflammatory conditions.
Declarations
Acknowledgement
References
Archives


News Updates